纳米孔测序和Shasta工具包实现11个人类基因组的从头组装
使用纳米孔长读序列重新组装人类基因组已有报道,但它使用了150,000 中央处理器小时和数周的挂钟时间。为了实现人类基因组的快速组装,我们提出了Shasta,一个从头开始的长读汇编器,以及名为MarginPolish和Helen的抛光算法。使用单个ProMethION纳米孔测序仪和我们的工具包,我们在9 d内重新组装了11个高度连续的人类基因组。我们在每个样本中使用三个流式细胞,获得了大约63倍的覆盖率,42kb的读取N50值和6.5kb的Reads>;100 kb的覆盖率。沙斯塔在单个商业计算节点上用不到6 h的时间完成了完整的单倍体人类基因组组装。MarginPolish和Helen仅用纳米孔就将单倍体组件抛光到99.9%以上的一致性(Phred Quality Score QV = 30)。邻近连接测序的增加使得所有11个基因组都可以接近染色体水平的支架。我们将我们的组装性能与现有的二倍体、单倍体和三倍体人类样本的组装方法进行了比较,并报告了优越的准确性和速度。
基于参考的方法,如GATK-1,可以从短读取序列中推断人类变异,但结果仅覆盖参考人类基因组组合2,3的~90%。这些方法对于参考基因组4的这一可映射部分的单核苷酸变异和短插入和缺失(INDel)是准确的。然而,对于从头基因组组合5,发现结构变异(SVS)6,7(包括大的INDel和碱基水平分辨的拷贝数变异)是困难的。但是,对于新的基因组组合5,发现结构变异(SVS)6、7(包括大的INDel和碱基水平分辨的拷贝数变异)是困难的。
第三代测序技术,包括连锁读数9、10、11和长读技术12、13,克服了用于基因组推断的短读测序的基本限制。除了越来越多地在参考引导方法2、14、15、16中使用之外,长阅读序列可以产生高度连续的从头开始基因组组件17。
牛津纳米孔技术公司(ONT)商业化的纳米孔测序对于从头组装基因组特别有用,因为它可以产生非常长的100千碱基(Kb)读数18的高产量。非常长的读数有望促进人类基因组中最具挑战性的区域的连续、不间断的组装,包括着丝粒卫星、末端着丝粒短臂、核糖体 阵列和最近的片段复制19、20、21。基于纳米孔测序的人类基因组的从头组装已被报道18。这一早期的工作需要53个微小的流式细胞,组装需要超过150,000 的中央处理器小时和数周的挂钟时间,这对于高通量的人类基因组测序工作是不可行的。
为了实现人类基因组的简单、廉价和快速从头组装,我们开发了一种用于纳米孔数据组装和抛光的工具包,其速度比最先进的方法快了几个数量级。我们结合使用纳米孔和邻近连接(HIC)测序9和我们的工具包,我们报告了人类基因组测序的改进,并减少了时间、劳动力和成本。
我们从1000个基因组计划22和瓶装基因组23样本中选择了11个低传代(6代)亲子三人组后代的人类细胞系进行测序。选择样本以最大化捕获的等位基因多样性(见方法)。
我们对这11个基因组进行了蛋白纳米孔测序和免疫测序。我们每个基因组使用三个流式细胞,每个流式细胞每隔20-24 小时接受一次核酸酶冲洗,这种冲洗去除了可能导致毛孔随着时间的推移而堵塞的长dna片段。在核酸酶冲洗后,每个流动细胞都收到一个相同样本的新文库。每个流式细胞总共进行两次核酸酶刷新,每个流式细胞总共获得三个测序文库。我们使用带有高精度触发器模型的Guppy v.2.3.5进行基本呼叫(参见方法)。
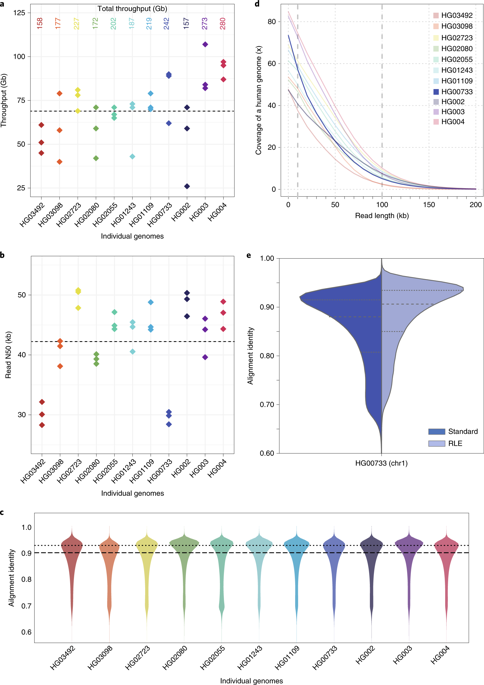
对全部11个基因组进行纳米孔测序,历时9 d,产生2.3Tb( Terabases,TB)序列。在这些测序过程中,我们并行运行了多达15个流式细胞。结果如图所示。纳米孔测序平均每个流动细胞产生69 千兆碱基(GB),每个基因组的总吞吐量在48× (15 8 GB)到85× (2 80 GB)覆盖范围内(图2)。(1A)。测序运行的读数N50值在28到51 kb(图2)之间。(1B)。(N50值是加权的中位数;它是集合中序列的长度,对于该集合,该长度或更大的所有序列的总和为集合总大小的50%。)。我们将纳米孔读数与人类参考基因组(GRCH38)进行了比对,并计算了它们的比对一致性以评估序列质量(参见方法)。我们观察到,中位数和模态比对一致性分别为90%和93%(F
A,对于11个样本,三个流动单元中的每一个的吞吐量以千兆基数为单位,总吞吐量最高。每个点都是一个流动单元。b、读取每个流动单元的N50值。每个点都是一个流动单元。C,与GRCh38对齐身份。a-c中的中点用虚线表示,c中的虚线是模式。每条线是由三个流动池组成的单个样品。D,基因组覆盖作为读取长度的函数。虚线表示覆盖范围为10和100 kb。例如,HG00733以深蓝色突出显示。每条线是由三个流动池组成的单个样品。E,标准和RLE读取的对齐标识。HG00733染色体1流式细胞1的数据如图所示(4.6 GB原始序列)。虚线表示四分位数。
在从纳米孔读数组装人类大小的基因组方面,Shasta被设计成比我们之前工作中使用的CANU汇编器快几个数量级,成本也低几个数量级。在大多数Shasta组装阶段,读数都是使用游程编码(RLE)24、25、26以均聚物压缩的形式存储的。在这种形式中,相同的连续碱基被折叠,并且存储碱基和重复计数。例如,GATTTACCA将表示为(GATACA,113121)。这种表示对均聚物游程长度中的错误不敏感,从而解决了牛津纳米孔读数12的主要错误模式。结果,由于读取错误而导致的组装噪声减少,并有助于显著更高的同一性比对(图。1E)。还使用读取的标记表示,其中每个读取在其游程表示中被表示为短k-MERS(标记表示)的预定的固定子集的出现序列。使用m个标记的连续出现(缺省为m个 = 4)作为最小散列特征,使用修改的最小散列27,28方案来寻找重叠读取的候选对。为所有候选对计算标记表示中的最佳对齐。标记表示中的路线计算非常有效,特别是在使用各种带状试探法时。创建标记图,其中每个顶点表示在若干读取的集合中发现对准的标记。标记图是经过一系列简化步骤后用来组装序列的。汇编器在具有大量内存的单台机器上运行(对于人工汇编,通常为1-2 TB)。所有数据结构都保存在内存中,除了读取的初始加载和汇编结果的最终输出之外,不会发生任何磁盘I/O。
我们将沙斯塔与三位同时代的汇编者进行了比较:Wtdbg2(参考文献2)。29),Flye 30和Canu 31。我们在两个二倍体人类样本HG00733和HG002以及一个单倍体人类样本CHM13的可用读取数据上运行了所有四个汇编程序。HG00733和HG002是我们收集的11个样本的一部分,CHM13的数据来自T2T联盟32。
CANU始终产生最连续的组装,样品HG00733、HG002和CHM13的重叠NG50值分别为40.6、32.3和79.5 Mb(图.。2A)。(NG50与N50相似,但只占估计基因组大小的50%。)。对于相同的样品,Flye是第二连续的,其重叠NG50值分别为25.225.935.3Mb Mb。沙斯塔紧随其后,其重叠群NG50值分别为21.1、20.2和41.1 Mb。Wtdbg2产生的邻接组装最少,重叠群NG50值分别为15.3、13.7和14.0 Mb。
a,NGx图显示重叠群长度分布。每条直线与虚线的交点是该部件的NG50。B,NGAx图显示了排列的重叠群长度的分布。每条水平线表示组件的对齐段,相对于GRCh38未被不一致或不可映射的序列打断。每条直线与虚线的交点是该部件的对齐NGA50。C,着丝粒外区域的组装不一致计数,节段性复制,对于HG002,已知SVS。d,总生成序列长度与总比对序列长度(对照GRCh38)。E.用于组合序列的平衡的基准级错误率。f,汇编程序的平均运行时间和成本(未显示CANU)。
相反,将样本与GRCh38对齐,并用Quast 33进行评估,与其他组装者相比,Shasta每个组装件的不一致(组装件相对于参考组装件包含断点的位置)减少4.2至6.5倍(补充表5)。打破这些相对于GRCh38的不一致和未对齐区域的组装,我们观察到连续的绝对变化要小得多(图4)。2B和补充表5)。然而,发现的分歧中有相当一部分可能反映了关于GRCh38的真实SVS。为了解决这个问题,我们对Y染色体、着丝粒、端着丝粒染色体臂、QH-区域和已知的最近节段性复制(所有这些都在SVS34,35中富集)内的折扣进行了折扣;在HG002的情况下,我们进一步
Shasta产生的碱基水平准确的组件最多(二倍体的平均平衡错误率为0.98%,单倍体的平均平衡错误率为0.54%),其次是Wtbdg2(二倍体的1.18%和单倍体的0.69%),CANU(二倍体的1.40%和单倍体的0.71%)和Flye(二倍体的1.64%和单倍体的2.21%)(图2)。2E,见方法和补充表12)。我们还计算了所有组件覆盖的区域的碱基水平准确性,并观察到与全基因组评估一致的结果(补充表13)。
Shasta、Wtdbg2和Flye在商业云上运行,使我们能够合理地比较它们的成本和运行时间(图2)。2E,请参见方法)。沙斯塔以平均每件样品70美元的成本完成每个组装平均需要5.25Mh( h)。相比之下,Wtdbg2耗时7.5倍,成本3.7倍,Flye耗时11.9倍,成本9.9倍。由于将CANU程序集移植到Amazon Web Services的预期成本和复杂性,CANU程序集在大型机构计算群集上运行,计算消耗高达19,000美元(估计),每个程序集需要大约4-5 d(方法,请参阅补充表14和15)。
为了评估使用Shasta进行SV表征的使用情况,我们创建了一个从Shasta组装图中提取假定杂合子SV的工作流程(方法)。从HG002的组装图中提取SVS,Indels的长度分布显示了已知反转录转座子长度的特征尖峰(补充图。1)。将这些SV与高置信度Giab SV集合进行比较,我们发现一致性很好,F1综合得分为0.68(补充表16)。
MHC区域由于其重复性和高度多态的性质37而很难使用短读取来解决,并且最近将长读取序列应用于该问题的努力已经显示出希望18、38。我们分析了CHM13和HG00733的组装,看看它们是否跨越了MHC区域。对于CHM13的单倍体组装,我们发现MHC在所有四个组合器的输出中完全被单个重叠群跨越,并且与GRCH38(图38)中提供的GL000251.2单倍群最为相似。3A,附图3A。2和补充表17)。在HG00733的二倍体组装中,两个重叠群横跨Shasta和Flye的大部分MHC,而Canu和Wtdbg2以一个重叠群跨越该区域(图.。3B和附图3。3)。然而,我们注意到,所有这些嵌合的二倍体组装导致的序列与任何单群(方法)并不十分相似。
用于CHM13和HG00733的未抛光的Shasta组件,包括HG00733三箱装母体和父体组件。灰色阴影区域是覆盖范围(与GRCh38对齐)降至20以下的区域。水平黑线表示重叠群中断。蓝色和绿色表示唯一对齐(分别向前和向后对齐),橙色表示多个对齐。
为了尝试解析HG00733的单倍型,我们使用三仓39根据可能的母系或父系将HG00733的读数分成两组,并组装单倍型(方法)。对于所有组装者和每个单倍型组装体,全局邻接性显著恶化(因为可用的读取数据覆盖率大约减半,而且,并不是所有的读取都可以分割),但是所产生的不一致计数减少(补充表18)。当使用单倍体三库组件时,MHC由母体单倍型的单个重叠群跨越(图4)。3C,附图3。4和补充表19),与GRCH38同源性高,并且与GL000255.1单倍型具有最大的邻接性和同一性。对于父本单倍型,低覆盖率会导致不连续(图3)。3D)将该区域划分为三个重叠群。
我们开发了一种基于深度神经网络的共识序列抛光流水线,旨在提高初始装配的基准级质量。该管道由两个模块组成:MarginPolish和用于纳米孔的均聚物编码长读纠错器(Helen)。MarginPolish在成对隐马尔可夫模型(Pair-HMM)上使用前向-后向算法的带状形式,从到程序集的每个读取器的RLE对齐生成成对对齐统计信息。根据这些统计数据,MarginPolish生成表示潜在可选局部组件的加权RLE偏序排列(POA)40图。MarginPolish使用此RLE POA迭代地细化程序集,然后输出最终摘要图供Helen使用。海伦使用多任务递归神经网络(RNN)41,该网络采用MarginPolish RLE POA图的权重来预测每个基因组位置的核苷酸碱基和游程长度。RNN利用上下文基因组特征和POA权重到正确碱基和游程长度的关联耦合来产生具有更高准确性的共识序列。
为了证明MarginPolish和Helen的有效性,我们将他们与国家-o进行了比较。
图4a和补充表20-22详细说明了使用Pomoxis在HG00733和CHM13 Shasta组件(参见方法)上执行的四种方法的错误率。对于二倍体的HG00733样本,MarginPolish和Helen的平衡错误率为0.388%(Phred质量分数QV = 24.12),相比之下,拉康和Medaka的错误率为0.455%(QV =23.42)。对于这两条抛光管道,这些误差中有相当一部分可能是由于真正的杂合变异造成的。对于单倍体CHM13,我们将比较限于由T2T联盟32提供的高度精选的X染色体序列。我们实现了0.064%(QV = 31.92)的均衡错误率,而Racon和Medaka的错误率为0.110%(QV = 为29.59)。
A,HG00733和CHM13上四种方法的均衡错误率。B,描述HG00733上的管线的四个步骤的给定真实运行长度(y轴)的预测运行长度(x轴)的行归一化热图。Guppy v.2.3.3是从3.7GBRLE序列中产生的。沙斯塔、马尔金·波利什和海伦是从与其各自对齐的整个集合中生成的。
..