人类梅毒群体的病毒目录显示慢性疾病协会
Warning: Can only detect less than 5000 characters
Warning: Can only detect less than 5000 characters
所有病毒基因组的基因组图(不包括严格的与远端条目的那些)沉积在Genbank和与Bioproject PrJNA573942相关的寄存号。根据NCBI指南,文件将在发布此手稿后发布。有关登录号码,请参阅DataSet S2。
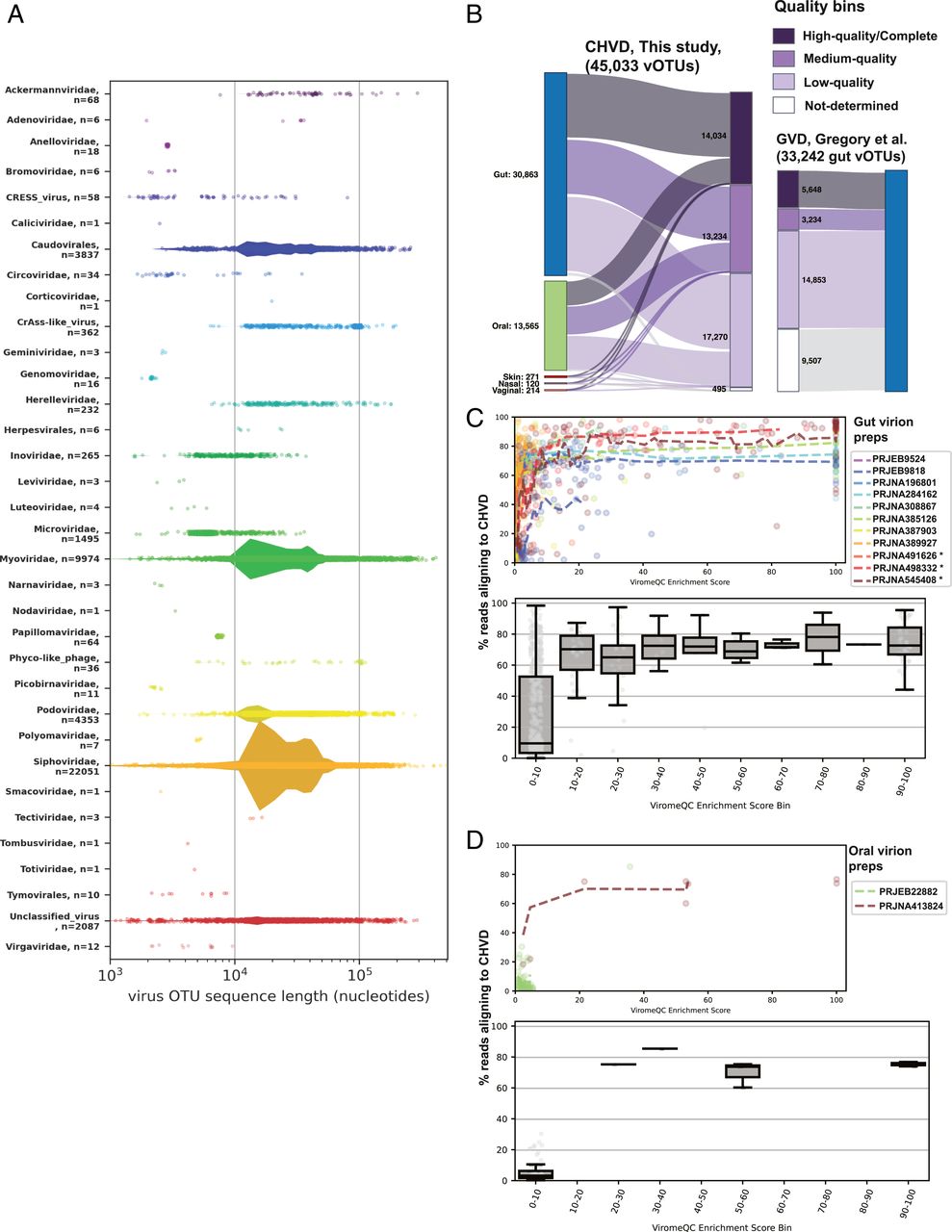
尚不清楚通过CHVD编目了多少人类病毒。解决这个问题的一种方法是查看物理富集病毒序列的数据集,并确定数据集中的读取部分是可识别的。从983个人粪便样品(代表11种不同的研究),该样品物理富含病毒粒子并进行核酸酶消化以去除不常用的核酸对准CHVD(图1c,上部)(数据集S5)。为了量化CHVD新兵从先前未分明的病毒赛准备中读取的良好,消除了CHVD生产中使用的样品,并计算了来自病毒评分的不同频率的中位数和狭窄的范围。该分析表明,与病毒浓缩评分的读取与CHVD尺度对齐的读数百分比。富含富集的(&lt 19 voromeqc得分)肠样品平均对齐约10%的读数,并且具有高富集分数(> 30)的样品平均对齐,其中许多样品实现了99%的对准。
虽然富含富集的病毒数据不适用于其他身体部位,但大约75%的读数在富集的病毒DNA富集的富集的口腔样品中是可分类的(图1D)(数据集S5)。
CRISPR间隔分析显示大多数码以及噬菌体竞争网络的候选主机。
许多细菌编码CRISPR-CAS系统,其含有从和用于入侵流动遗传元件,尤其是噬菌体(62)的粘隙间隔物阵列。将细菌CRISPR间隔序列与噬菌体基因组匹配是确定细菌谱系是否先前暴露于特定噬菌体的一种方式。从细菌基因组编目和噬菌体/宿主匹配管道的优化对CRISPR间隔物的进展允许将该项目中发现的大部分噬菌体的关联与细菌宿主(http://crispr.genome.ulaval.ca//)(63)。具体而言,45,033病毒序列的31,259份与已知的细菌或多个细菌至少有一个Cr Clasp间隔匹配,与CHVD序列中的独特基因座匹配369,465个总垫片(数据集S2)。 CRISPR间隔密度在不同的细菌征集中有显着变化(图2A)。例如,确认双歧杆菌属的成员具有相对较大和多样化的克隆间隔子文库(64),而梭菌,藻藻藻菌和百叶菌通常只编码一个或少数每种噬菌体的间隔物。
噬菌体本身可以编码CRISPR阵列,并且一些噬菌体具有完整和功能的CRISPR-CAS系统(22,65)。这些CRISPR组件可以针对主机防御以及其他竞争对手的鼠标(66)。在CHVD中的噬菌体序列中,从203个噬菌体的基因组中检测到1,971个CRISPR间隔物。其中,799个间隔物靶向了2,036个其他噬菌体,表明人体梅毒(图2b)中的复杂噬菌体噬菌体竞争网络(从https://zenodo.org/record/449884下载cytoscape文件)(56)。 CRISPR-编码噬菌体和它们的目标噬菌体的细菌宿主池应相同。因此,记录了对噬菌体噬菌体对的细菌CRISPR间隔匹配,并且当可以针对CRISPR编码的噬菌体和靶噬菌体测定细菌宿主时,该细菌属相同,对于85.1%的对(数据集S6)。
通过这种病毒库和来自人类微生物组项目的大型抽样努力(34,67),“哪些病毒是最常见的是最丰富的”的问题可以比以前更自信地回答。应该指出的是,人类微生物组项目数据从18至40岁之间的健康美国人收集,这里的结论可能不完全普遍存在于其他人群。
在没有实验验证的情况下精确确定综合性染色体序列与宿主染色体序列之间的边界往往有挑战性,使病毒量化在全基因组霰弹枪(WGS)数据集具有挑战性。我们的初步分析显示,在病毒OTU Contig中包含侧翼宿主序列的几百个核苷酸可以大大扭曲丰富的丰富测量,因为无感染的宿主细菌序列的测量。因此,我们进行了更严格的分析,其中通过最后的病毒标志基因从第一公认的病毒标志基因修剪了折叠。在去除一些病毒序列的同时,该方法保留了病毒基因组最不可磨灭的序列,同时所有但确保不会保留没有细菌染色体。我们将这些更严格的单位称为“病毒核心”(从https://zenodo.org/record/4498884下载)(56)。
数据从SRA下载,分析了数百名六位体位(前鼻腔,口腔粘膜,后穹窿,舌窝,Suprictival斑块和肠道[粪便])。然后将读取与更严格的病毒核心数据库对齐。作为给定病毒OTU的相对丰度的代理,针对每个序列计算父数据集(RPKM)中每百万读数的病毒基因组每千碱基的平均读数的平均数量(图3,SI附录。S2和S3和数据集S2)。病毒患病率被确定为样品的比例和gt; 0.1 rpkm。最常见的病毒OTUS计算为(平均RPKM次普及率)。图3的右图。图3示出了基于CRISPL间隔目标信息的前30个最常见的vorus中的每一个的推断宿主。大多数最常见的大量病毒似乎感染常见的细菌族菌科的成员,这在人体肠道中通常丰富。此外,尽管与过去的研究相比,尽管对可对病毒序列进行了大幅增加,但是在人体肠道生态系统(12)中,粗糙的噬菌体高度丰富的观察似乎似乎非常好。
该数据表明,具有高丰度(RPKM)的肠道病毒OTU流行的有趣分叉。虽然有些病毒Otus,例如podoviridae sp。 ctBGm1和长尾噬菌体科属。 ctrxw1,存在于几乎所有的样品和具有≥280℃的平均丰度; 10 RPKM,或许较普遍存在的细菌谱系的噬菌体。其他,包括所有显示的粗糙的病毒和myoviridae sp。 CTNBA1在大多数样品中不存在或低丰度,但在少数样本中非常丰富。后一组可以代表定期进行大量复制爆发的病毒,或者在某些个人中组成统治生物的病毒,但不是其他人。
正如预期的那样,大多数病毒OTU在只有一个车身部位普遍存在,但是186个“COSOMOPOLITAN”OTUS的患病率> 0.2(即20%
......