超导量子比特计算机上的Hartree-Fock
2020-08-28 17:00:54
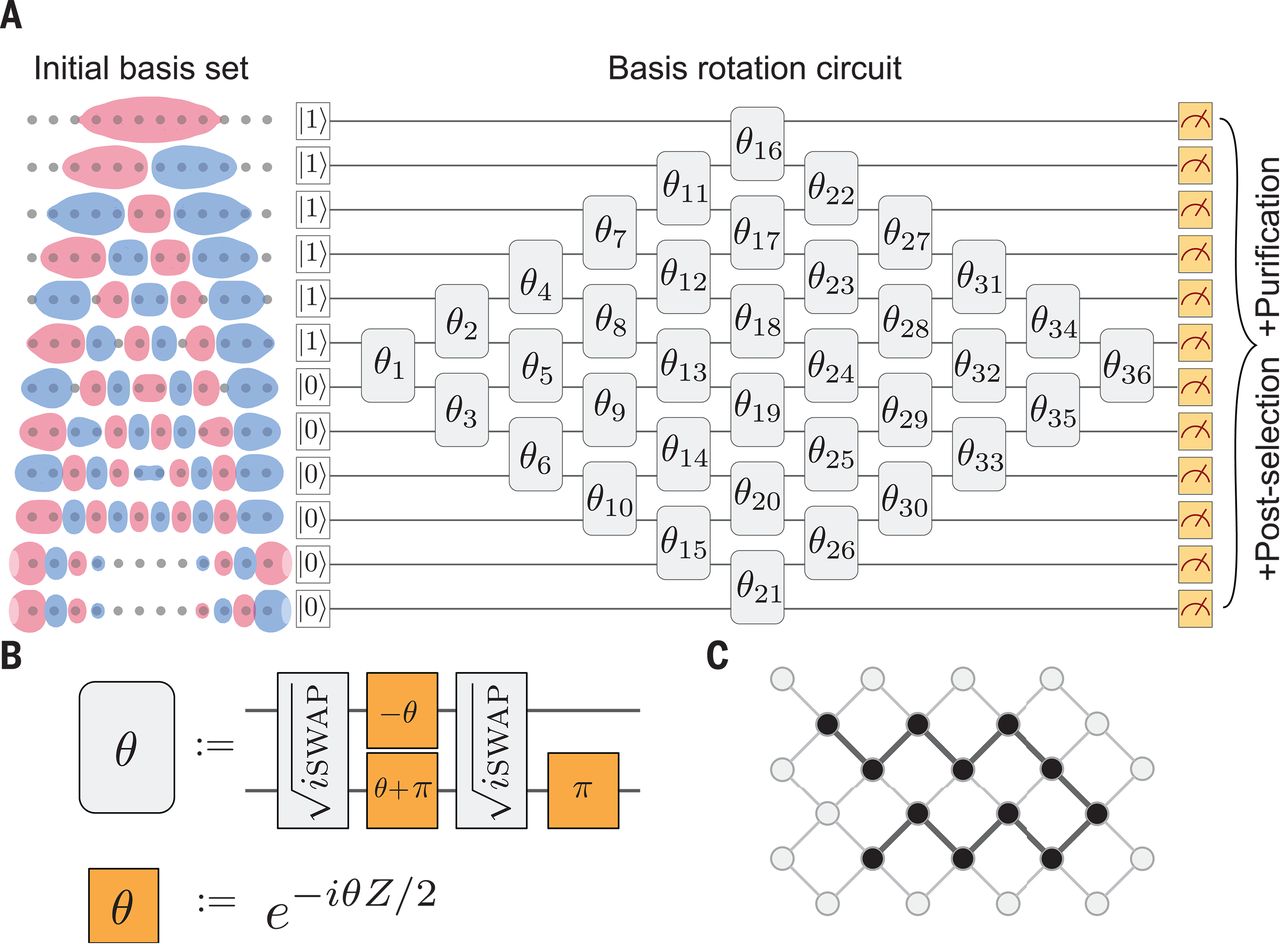
精确的电子结构计算被认为是量子计算最令人期待的应用之一,它将给理论化学和其他相关领域带来革命性的变化。使用Google Sycamore量子处理器,Google AI Quantum和合作者对两个中等规模的化学问题进行了变分量子本征解算器(VQE)模拟:氢链的结合能(长达H12)和双氮烯的异构化机理(见袁的观点)。这些模拟是在多达12个量子比特上进行的,涉及多达72个两量子比特的门,并表明当VQE与误差缓解策略相结合时,有可能达到化学精度。所提出的VQE算法的关键构件有可能扩展到不能经典模拟的更大的系统。科学,这一问题,第[1084]页[1];另见第[1054]页[2]费米子系统的模拟是量子计算最期待的应用之一。我们用多达12个量子比特进行了几个量子化学的量子模拟,包括模拟双氮烯的异构化机理。我们还展示了基于N表示能力的错误缓解策略,显著提高了实验的有效保真度。我们的参数化的ANSATZ电路实现了非相互作用费米子演化的吉文斯旋转方法,我们对其进行了变分优化,以制备Hartree-Fock波函数。这个无处不在的算法原语在经典上易于模拟,但仍然在计算基础上产生高度纠缠态,这使得我们能够评估我们硬件的性能,并为扩大关联量子化学模拟奠定基础。[1]:/Lookup/doi/10.1126/cience.abb9811[2]:/lookup/doi/10.1126/cience.abd3880